Amyotrophie spinale proximale : comprendre, diagnostiquer et vivre avec la maladie

Qu’est-ce que l’amyotrophie spinale proximale ?
Décrite pour la première fois à la fin du XIXe siècle par le neurologue Autrichien Guido Werdnig et le neurologue allemand Johann Hoffmann, l’amyotrophie spinale proximale (ou SMA pour Spinal Muscular Amyotrophy) est une maladie neuromusculaire rare qui concerne principalement les enfants mais aussi les adultes.
Concrètement, l’amyotrophie spinale proximale, c’est quoi ?
Cette pathologie vient changer la façon dont le corps bouge. Elle touche les motoneurones, ces cellules nerveuses essentielles à notre corps pour contrôler les mouvements volontaires. Ce sont ces cellules qui, depuis la moelle épinière, donnent les “ordres” du cerveau aux muscles. Parfois les motoneurones présentent des anomalies génétiques graves et les muscles ne reçoivent plus le message moteur. En fonction de l’avancée de la maladie, les muscles (surtout ceux proches du tronc : les épaules, les cuisses et le dos – c’est pour cela qu’on appelle ça “proximale”) perdent de leur force, faute de stimulation.
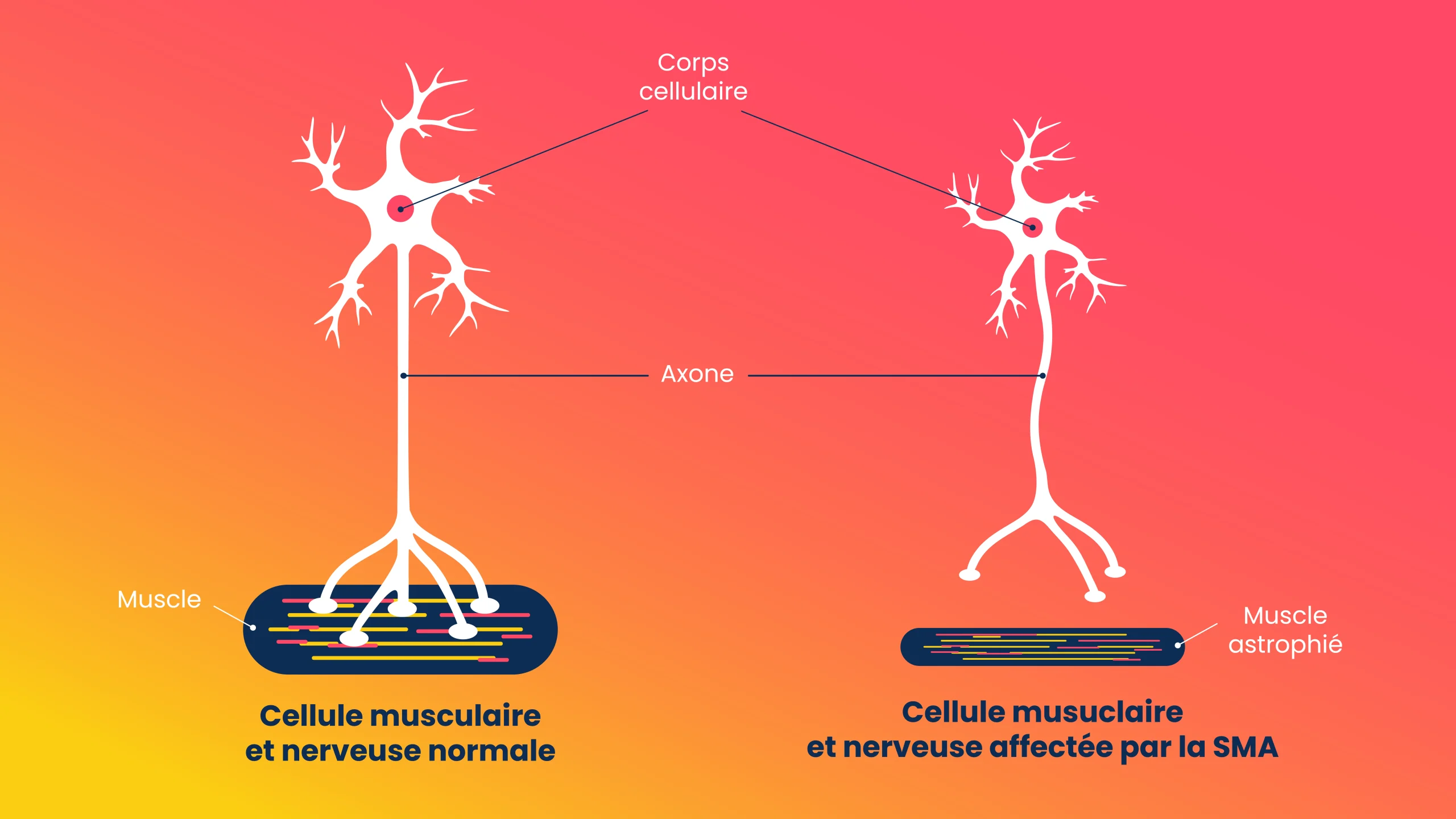
On appelle ça une amyotrophie ou en d’autres termes : une fonte musculaire.
Mais qu’est-ce qu’une amyotrophie au juste ? Et bien c’est une diminution du volume d’un muscle. Il faut imaginer un ballon de baudruche qui se dégonfle. En fait, le muscle “fond” parce qu’il a moins de fibres musculaires qui le composent. Moins de fibres, c’est moins de force et donc une capacité moindre à faire des mouvements. Cette fonte peut affecter un seul muscle, plusieurs ou même le corps entier.
Dans certains cas, l’amyotrophie apparait seulement lorsqu’un muscle n’est pas assez sollicité (comme une personne alitée ou après avoir porté un plâtre). Et dans d’autres cas, elle est liée à des maladies génétiques qui affectent directement le muscle (myopathies) ou les nerfs (comme l’amyotrophie spinale proximale).
La SMA apparaît dès la naissance ou dans les premiers mois. Elle peut survenir aussi chez l’enfant et plus rarement chez l’adolescent ou l’âge adulte. Suivant le moment où elle se manifeste, elle limite certains gestes du quotidien : se tenir assis, marcher, lever les bras, garder l’équilibre voire même respirer dans les formes plus sévères. C’est seulement le corps qui se fatigue petit à petit, pas l’esprit.
Qui est concerné ? Épidémiologie et groupes à risque
L’amyotrophie spinale proximale fait partie des maladies neuromusculaires rares. Elle est transmise génétiquement de manière autosomique récessive (il faut que les deux parents donnent le gène qui est défectueux).
Cette pathologie touche le genre féminin et masculin (à contrario de la dystrophie musculaire de Duchenne, une autre maladie neuromusculaire qui est essentiellement présente chez les garçons). Environ 2500 personnes vivent avec la SMA en France, tous types (0, I, II, III, IV) confondus (+120 nouveaux cas par an). Ces 4 types se divisent en fonction de l’âge d’apparition et de gravité : la prévalence globale est estimée à 1 cas pour 30 000 naissances d’après Orphanet. Les plus jeunes sont les plus impactés (principalement les nourrissons et les enfants en bas âge).
La SMA de type 0 : extrêmement rare qui est présente durant la période prénatale. On remarque une faible activité fœtale pendant la grossesse due à une faiblesse musculaire.
La SMA de type I : c’est la forme la plus sévère et la plus fréquente (environ 60% des cas). Elle se manifeste lors des 6 premiers mois de vie avec une faiblesse musculaire importante.
La SMA de type II : apparaît entre 6 et 18 mois. Elle représente 30% des cas et est moins sévère que le type I. Dans cette forme, les enfants peuvent encore s’asseoir sans aide mais pour la plupart ils ne marchent pas.
La SMA de type III : elle survient plus tard dans l’enfance, entre 2 et 12 ans. C’est 10% des cas. Les personnes qui vivent avec ce type de SMA continuent de marcher mais avec des difficultés au fil du temps.
La SMA de type IV : c’est la forme “adulte” de la SMA et la plus rare, seulement 1% des cas. Ce type a une évolution lente et à peu d’impact sur l’espérance de vie.
Ces chiffres font de l’amyotrophie spinale proximale une maladie, dite, rare. En Europe, ces maladies rares concernent 1 personne sur 2000.
Les différentes formes cliniques de la SMA
Type 1 : forme infantile sévère
L’amyotrophie spinale proximale de type I est la forme la plus fréquente et la plus sévère de tous les types de SMA. Elle débute généralement avant l’âge de 6 mois. Elle se caractérise par une hypotonie (une grande faiblesse musculaire majeure) et une mobilité très réduite, principalement au niveau des membres inférieurs. Par conséquent, les bébés prennent souvent une posture de “grenouille” quand ils sont allongés sur le dos.
On observe aussi un balancement thoraco-abdominal pendant la respiration. C’est lorsque le thorax qui se rétrécit et ventre qui ressort à l’inspiration, signe de difficultés respiratoires.
D’autres symptômes sont à surveiller : les fasciculations (des petits frémissements) de la langue et une disparition des réflexes musculaires “normaux” ou encore des incapacités à déglutir correctement. Sans traitement adapté, les nourrissons qui vivent avec la SMA de type I, maladie qui évolue très rapidement, décèdent vers 2 ans.
Type 2 : forme intermédiaire
Cette forme intermédiaire de la SMA de type II apparaît entre 6 et 18 mois. Les enfants, avec ce type, s’assoient seuls mais ne peuvent pas marcher sans une aide technique (fauteuil roulant ou aide à la marche).
On observe souvent :
- un enroulement en bas du dos quand ils sont assis (on appelle ça une cyphose asthénique)
- un retard dans les mouvements (se retourner, utiliser ses mains de manière coordonnée, etc.)
- une fonte musculaire (amyotrophie)
- des tremblements dans les mains
- des fasciculations
Progressivement des difficultés compliquent le quotidien, comme la scoliose, des problèmes orthopédiques ou encore des difficultés respiratoires ou alimentaires. Un accompagnement socio-médical améliore la qualité de vie de la personne en situation de handicap. Avec une SMA de type II, les personnes atteignent l’âge adulte bien que des complications surviennent avec le temps. L’espérance de vie est en moyenne de 30 à 50 ans.
Type 3 : forme juvénile
La forme de type III de l’amyotrophie spinale proximale apparaît chez les enfants et les adolescents qui ont commencé à marcher après 18 mois (un nourrisson sans SMA de type III apprend la marche entre 10 et 18 mois).
Ces enfants et adolescents marchent de façon autonome mais des troubles moteurs progressifs apparaissent peu à peu. Ils ont une marche dandinante souvent sur la pointe des pieds ou avec des pieds à plat (et ça c’est pour la plupart des cas, des signes d’un déficit musculaire proximal = faiblesse ressentie dans les muscles proches du tronc).
Des mouvements du quotidien comme monter des escaliers, courir ou se relever du sol deviennent alors compliqués.
Dans ce type de SMA, on observe, comme dans la SMA de type II, une amyotrophie, des tremblements des mains et parfois des fasciculations. Les réflexes ostéo-tendineux (les réflexes que le médecin teste en “cognant” très légèrement un marteau à réflexe sur un genou) sont souvent diminués ou absents.
Marcher devient, dans certains cas, compliqué. Mais il est important de rappeler que l’espérance de vie reste normale.
Des symptômes supplémentaires peuvent intervenir : scoliose, douleurs musculaires ou troubles respiratoires légers.
Type 4 : forme adulte
Pour rappel, c’est la forme la plus rare des types de SMA. Elle se déclare à l’âge adulte, entre 20 et 30 ans et évolue lentement : ce qui peut la rendre confondante avec d’autres maladies neurodégénératives.
Ces premiers symptômes sont difficiles à détecter, il y a d’abord une faiblesse musculaire au niveau des jambes et des hanches qui s’étend ensuite aux épaules puis aux bras.
Le reste des symptômes sont pratiquement les mêmes que la SMA de type III :
- une démarche qui devient dandinante
- des tremblements des doigts
- des fasciculations
- ou encore une hypertrophie des mollets (= une augmentation anormale de leur volume liée à la maladie)
À noter que cette pathologie ne touche pas les capacités intellectuelles, elles restent totalement préservées.
Proportion des cas totaux
Type de SMA | Âge d’apparition | Épidémiologie, nombre de cas annuel | Proportions des cas totaux | Particularités épidémiologiques | Type 1 (SMA1) | Avant 6 mois (nourrisson) | Environ 7 200/ 12 000, soit 33 / 663 000* de cas par an | Environ 60 % des cas | Forme la plus fréquente et sévère, apparition très précoce, mortalité infantile élevée sans traitement |
|---|---|---|---|---|
Type 2 (SMA2) | Entre 7 et 18 mois | Environ 3 600/ 12 000, soit 17/ 663 000 de cas par an | Environ 30 % des cas | Début dans la petite enfance, faiblesse musculaire empêchant la marche sans aide, atteinte respiratoire possible, légère prédominance masculine |
Type 3 (SMA 3) | Entre 18 mois et l’âge adulte (souvent vers 3 ans) | Environ 1 200/ 12 000, soit 6 / 663 000 de cas par an
| Environ 10 % des cas
| Forme juvénile ou adulte jeune, évolution lente, marche possible longtemps, chutes fréquentes, troubles moteurs progressifs
|
Type 4 (SMA4) | À l’âge adulte (deuxième ou troisième décennie) | Environ 120/ 12 000, soit 0,55 / 663 000 de cas par an | Environ 1 % des cas | Forme adulte, évolution très lente, faiblesse musculaire proximale légère, espérance de vie normale, symptômes proches de la SMA3 mais plus légers |
* 663 000 est le nombre de nouvelles naissances enregistrées en France en 2024
Origine et mécanisme génétique de la maladie
La SMA est causée par une mutation du gène SMN1, c’est un gène qui code la protéine SMN (Survival Motor Neuron) indispensable pour les motoneurones et leurs survies.
Les deux copies du gène SMN1 sont absentes ou non fonctionnelles dans environ 95% des cas, ce qui paralyse la production de cette protéine primordiale à ces cellules nerveuses.
Mais il existe le gène SMN2, le “cousin” du gène SMN1, presque identique. Il y a une petite différence dans sa séquence (c’est-à-dire dans l’ordre des “lettres” qui composent son code génétique), il produit une version tronquée et moins efficace de la protéine SMN.
Le nombre de copies de SMN2 change d’un individu à l’autre : plus il y a de copies, plus la maladie est freinée car elles prennent le relais du gène SMN1.
Par exemple, si un nourrisson a 3 ou 4 copies de gène SMN2, il aura une forme plus légère que celui qui n’en a qu’une seule. La présence de SMN2 agit comme un petit “coussin” : elle modère la gravité de la maladie mais elle ne la guérit pas.
💡
Imaginons que le gène SMN1 est le cuisinier principal d’un restaurant. Quand il est absent, c’est le gène SMN2 qui le remplace. Mais SMN2 est un apprenti cuisinier en phase d’apprentissage : il fait ce qu’il peut avec les connaissances qu’il a. Et plus on a d’apprentis en cuisine, plus le repas (ici la protéine) a de la chance d’être à peu près réussi.
Reconnaître les signes d’alerte : diagnostic clinique et génétique
Il•elle réalise également un test génétique, c’est très important afin de détecter une mutation ou une délétion (perte de matériel génétique sur un chromosome) du gène SMN1.
🐙
Le point vocab’ du poulpe : le test génétique
C’est un test qui permet de confirmer la mutation du gène. C’est crucial pour établir un diagnostic précis.
Un diagnostic précoce est essentiel : il existe certains traitements (comme le nusinersen ou le thérapies géniques) qui sont plus efficaces avant que la personne n’ait perdu ses motoneurones.
Des études montrent que sans un traitement, un nourrisson de type I perd jusqu’à 90% de ses motoneurones avant ses 6 mois. C’est pourquoi un dépistage néonatal (des médecins prélèvent deux gouttes de sang sur le talon du nouveau-né) se met en place dans des recherches cliniques en France avec un délai rapide entre le prélèvement, la confirmation du diagnostic et le début du traitement.
Traitements actuels et avancées thérapeutiques
Thérapie génique qui utilise un vecteur viral AAV9 pour apporter une version fonctionnelle du gène SMN1 directement aux motoneurones
Prise quotidienne par voie orale dès 2 mois. C’est le premier traitement oral approuvé aux États-Unis
Amélioration motrice notée dans les types II et III prouvée par l’essai SUNFISH
Nusinersen (Spinraza)
Mécanisme : agit sur le gène SMN2 pour qu’il produise plus de protéine SMN
Administration : injection directement dans la moelle épinière avec 4 doses initiales en 2 mois puis une dose tous les 4 mois
Efficacité : Amélioration et stabilisation de la fonction motrice (notamment chez les formes précoces de type I et des progrès dans les types II et III)
Limites : N’atteint pas le traitement moteur maximal. Le traitement est invasif, répété et coûteux
Onasemnogene abeparvovec (Zolgensma®)
Mécanisme : thérapie génique qui utilise un vecteur viral AAV9 pour apporter une version fonctionnelle du gène SMN1 directement aux motoneurones
Administration : Perfusion intraveineuse, avant l’âge de 2 ans. Un traitement pour calmer le système immunitaire est nécessaire afin d’éviter des problèmes de foie
Efficacité : Amélioration des fonctions motrices et survie prolongée parfois jusqu’à 5 ans après traitement
Limites : Efficacité décroissante avec l’âge, contre-indications en cas d’anticorps anti-AAV9 et risque d’hépatotoxicité
Risdiplam (Evrysdi®)
Mécanisme : Médicament à avaler qui agit sur le gène SMN2 pour produire plus de protéine SMN dans tout le corps
Administration : Prise quotidienne par voie orale dès 2 mois. C’est le premier traitement oral approuvé aux États-Unis
Efficacité : Amélioration motrice notée dans les types II et III prouvée par l’essai SUNFISH
Limites : Effets secondaires digestifs et cutanés
La recherche continue pour aller encore plus loin dans l’efficacité et s’adapter à encore plus de personnes :
- La thérapie génique améliorée : des chercheur•euses travaillent sur des nouveaux vecteurs (comme des véhicules qui conduisent le gène SMN1 dans le corps) pour que cela marche sur des personnes âgées.
- L’édition du génome (CRISPR/Cas 9) : une nouvelle technologie qui a pour but de modifier directement la mutation du gène SMN1 à l’intérieur des cellules. Le but ? Réparer directement “à la racine” sans avoir à stimuler le gène SMN2, comme les traitements d’aujourd’hui.
- Les biothérapies ciblées : de nouveaux médicaments qui protègent les motoneurones et/ou renforcent les muscles. Certaines de leurs molécules visent aussi à corriger l’épissage (le montage de l’ARN qui permet de fabriquer la protéine complète),comme le Spinraza ou le Risdiplam mais en beaucoup plus puissant.
Ces 3 pistes sont encore au stade d’étude et de test, elles ne sont pas disponibles en pratique mais elles ouvrent des nouvelles perspectives qui pourraient aider à mieux comprendre la SMA et à perfectionner la prise en charge des personnes.
Une prise en charge globale et pluridisciplinaire
Quand on vit avec une SMA, il faut être accompagné par des professionnel•les de santé :
- La rééducation fonctionnelle et kinésithérapie motrice : en lien avec un•e kinésithérapeute spécialisé dans les maladies neuromusculaires, il•elle propose des exercices adaptés pour prévenir les raideurs, conserver au maximum le mouvement et améliorer l’autonomie et la qualité de vie au quotidien.
- La kinésithérapie respiratoire : pour aider à sortir les sécrétions bronchiques avec des techniques de drainage, d’aide à la toux ou d’appareils comme l’in-exsufflateur.
- L’orthophonie : l’orthophoniste accompagne les personnes qui vivent avec une SMA, dans les troubles de la déglutition et de la parole.
- La nutrition : un•e nutritionniste prévient de la malnutrition (assez courant avec la SMA). En cas de problèmes alimentaires sévères, une gastrostomie est envisagée.
- L’ergothérapie : il•elle propose des aides techniques pour améliorer le quotidien (fauteuil roulant, bras robotique pour handicap…).
- Soutien scolaire : avec la fatigue d’écrire, la posture, etc., les équipes éducatives mettent en place différents outils : ordinateur, temps supplémentaire ou encore l’aménagement de l’espace de la classe.
- Un accompagnement psychologique et social : une•e psychologue peut accompagner la personne qui vit avec la SMA mais aussi son entourage, pour gérer l’aspect plus émotionnel et sur comment gérer la maladie. Les assistant•es sociaux vont aider pour l’accès au droit et/ou au soutien de la vie quotidienne.
Neurologues, pneumologues, rééducateur•rices, orthophonistes, nutritionnistes, psychologues… sont organisée avec ce qu’on appelle un coordinateur médical et d’un point de contact (souvent ce sont des infirmier•ères référent•e, des associations ou un•e coordinateur•rice de centre de soin).
Le fait d’être coordonné de la sorte permet d’avoir un suivi fluide, des bilans réguliers et des ajustements selon l’évolution de l’enfant ou de l’adulte.
Vivre avec une amyotrophie spinale : autonomie, inclusion et innovations
Est-ce que l’amyotrophie spinale empêche de vivre pleinement ?
Spoiler : Non !
Avec des solutions technologiques bien pensées et adaptées qui viennent soutenir le quotidien et compenser la perte de force musculaire, les personnes qui vivent avec une SMA pourront continuer d’aller dans leur restaurant préféré ou même d’aller au musée !
Parmi les solutions technologiques on retrouve les exosquelettes. L’exosquelette est un équipement articulé fixé sur un membre pour faciliter les mouvements. Il est conçu pour assister dans les tâches du quotidien, augmenter la force, la mobilité et aussi l’endurance de la personne équipée.
Le projet espagnol Marsi Bionics, soutenu par l’Union Européenne, développe un exosquelette pour que les enfants avec une maladies neuromusculaires puissent se tenir debout.
Nous pouvons également parler des assistants robotiques pour la mobilité des bras comme notre dispositif médical, l’ORTHOPUS Supporter.
L’ORTHOPUS Supporter accompagne les mouvements verticaux et rend le bras plus léger.
Il fonctionne avec deux modes, FIXE & LIBRE, selon les besoins et les préférences de l’utilisateur•rice.
- Le mode FIXE qui accompagne le mouvement sur une position choisie afin d’avoir plus de stabilité. C’est le mode parfait pour se brosser les dents !
- Le mode LIBRE soutient le bras comme s’il flottait dans l’eau. Les gestes sont fluides et l’utilisateur•rice gagne en autonomie au quotidien.
Ces modes compensent la mobilité selon les capacités de chacun•e.
L’ORTHOPUS Supporter se branche à la batterie du fauteuil roulant électrique (mais il ne la vide pas ! C’est fait pour, au contraire ! 😉) et sur prise secteur si la personne souhaite l’utiliser seulement sur table (pour manger ou faire un puzzle).
Comme ORTHOPUS, d’autres solutions misent également sur l’open source (technologie partagé librement), ce qui offre la possibilité à des personnes qui vivent avec une maladie neuromusculaire, des makers ou ingénieur•e de co-concevoir leurs aides sur mesure.
Dans la même idée que notre bras robotique open source, nous pouvons parler de e-NABLE ou Open Bionics avec leurs bras imprimés en 3D qui sont accessibles et adaptables selon les besoins.
Ces modes de compensation doivent non pas seulement être fonctionnels, ils doivent aussi être désirables, personnalisables et inclusifs.
Comme l’ORTHOPUS Supporter, de plus en plus de solutions technologiques incluent les utilisateur•rices dès la conception (co-design) pour s’assurer que ces dispositifs s’adaptent réellement à leur vie. Ce design participatif est aujourd’hui un levier d’innovation et technologique.
Accompagner les proches : rôle de l'entourage et des aidants
Les proches de la personne avec une amyotrophie spinale proximale sont impactés psychologiquement et émotionnellement aussi. L’annonce du diagnostic, les soins à organiser, les ajustements de vie (déplacements, logement, travail…) engendre du stress, un sentiment d’isolement et/ou d’épuisement. Pour certain•es, c’est un “trop-plein mental et logistique” où les rôles de parent, d’aidant, de conjoint ou même d’ami se mélangent.
Pour aider ces aidant•es, il existe des solutions :
- Des formations pour apprendre les bons gestes, prendre confiance, comprendre la maladie, comment s’y prendre avec son proche, existent, comme les formations de l’Association Françaises des Aidants.
- Des groupes de parole : pour échanger avec d’autres aidant•es et des professionnel•les comme ceux organisés par les Unions Départementales des Associations Familiales ou les associations d’aidants.
- Des ressources associatives qui accompagnent au niveau social, juridique et/ou psychologique.
- Des dispositifs comme le congé proche aidant ou les haltes-répits pour souffler quelques heures.
Et ce qui est important c’est de co-construire l’environnement quotidien avec la personne concernée. Il faut comprendre ses besoins et ses préférences, aménager le quotidien.
Questions fréquentes autour de la SMA
Est-ce que l’amyotrophie se soigne ?
L’amyotrophie en tant que telle n’est pas une maladie (ne pas confondre avec la SMA ou la SLA qui sont elles, des maladies), c’est simplement une perte de masse musculaire (ou un symptôme de certaines maladies neuromusculaires). L’amyotrophie peut parfois être partiellement ou totalement soignée.
L’amyotrophie spinale proximale, elle, n’a pas de guérison complète mais des traitements ralentissent sa progression, améliore la force musculaire et la qualité de vie.
L’amyotrophie spinale est-elle une maladie rare ?
Quels sont les symptômes de l’amyotrophie spinale ?
Les symptômes ne sont pas les mêmes car il existe plusieurs types de SMA (0, I, II, III, IV). Généralement tous les types ont ces symptômes :
- Faiblesse musculaire progressive
- Perte de tonus (hypotonie)
- Réflexes diminués
- Fasciculations
- Difficultés motrices
- Complications respiratoires et/ou nutritionnelles
Qu’est-ce que l’atteinte proximale ?
Sources :
- https://www.chu-lyon.fr/amyotrophie-spinale-sma
- https://biogenlinc.fr/fr/sma/signes-and-symptomes/
- https://www.has-sante.fr/upload/docs/application/pdf/2023-07/note_de_cadrage_depistage_neonatal_amyotrophie_spinale_vf.pdf
- https://www.plemara.fr/maladies-rares/definition/#:~:text=D’apr%C3%A8s%20la%20d%C3%A9finition%20de,%C3%A0%201%20personne%20sur%202500
- https://www.orpha.net/fr/disease/detail/83330
- https://pubmed.ncbi.nlm.nih.gov/33090613/
- https://www.myobase.org/doc_num.php?explnum_id=23921&utm_
- https://www.sciencedirect.com/science/article/pii/S1773035X25762772?utm_
- https://fr.wikipedia.org/wiki/Amyotrophie_spinale?utm_
- https://www.roche.fr/articles/amyotrophie-spinale-therapies-innovantes?utm_
- https://www.sante-sur-le-net.com/maladies/maladies-rares-genetiques/amyotrophie-spinale-type-1/?utm_
- https://www.institut-myologie.org/2020/10/12/diagnostiquer-la-sma-avec-les-techniques-de-sequencage-de-nouvelle-generation/?utm_
- https://www.afm-telethon.fr/fr/fiches-maladies/amyotrophie-spinale-proximale?utm_
- https://www.myobase.org/index.php?id=71138&lvl=notice_display&utm_
- https://www.curesma.org/wp-content/uploads/2022/10/WEB-SMA-Diagnosis-from-Newborn_Screening_French.pdf?utm_
- https://www.afm-telethon.fr/fr/actualites/les-aav-vecteurs-de-therapie-genique-de-choix-dans-les-maladies-neuromusculaires
- https://www.ema.europa.eu/en/medicines/human/EPAR/evrysdi
- https://www.afm-telethon.fr/fr/actualites/sma-le-risdiplam-evrysdi-autorise-aux-etats-unis#:~:text=La%20FDA%20approuve%20le%20risdiplam,type%201%2C%202%20et%203.
- https://www.embopress.org/doi/full/10.1038/s44321-024-00036-y?utm_
- https://www.reddit.com/r/CRISPR/comments/16rs1ox/how_far_along_has_crispr_come_are_they_using_this/?utm_
- https://link.springer.com/article/10.1007/s40263-022-00941-1?utm_
- https://link.springer.com/article/10.1007/s40263-022-00941-1?utm_
- https://fr.wikipedia.org/wiki/Nusinersen?utm_
- https://orthopedicreviews.openmedicalpublishing.org/article/25579-risdiplam-for-the-use-of-spinal-muscular-atrophy?utm_
- https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213535Orig1s000MedR.pdf?utm_
- https://www.afm-telethon.fr/fr/fiches-maladies/amyotrophie-spinale-proximale-liee-smn1?utm_
- https://www.orpha.net/pdfs/data/patho/Han/Int/fr/AmyotrophieSpinaleProximaleType1_FR_fr_HAN_ORPHA83330.pdf?
- https://assets.roche.com/f/173873/x/4f0801cb3c/amyotrophie_spinale_infantile_-_pnds_20210402.pdf?utm_
- https://www.sciencedirect.com/science/article/pii/S0960896617312907?utm_
- https://www.treat-nmd.org/wp-content/uploads/2023/02/SMA-In-English-1.pdf?utm_
- https://www.orpha.net/pdfs/data/patho/Pub/fr/AmyotrophieSpinaleProximale-FRfrPub633v01.pdf?utm_
- https://www.saintluc.be/fr/hepato-gastro-enterologie-documentation-gastrostomie#:~:text=Qu’est%2Dce%20qu’,recevoir%20l’apport%20%C3%A9nerg%C3%A9tique%20n%C3%A9cessaire.
- https://cordis.europa.eu/article/id/430453-wearable-robots-provide-extra-muscle-for-children/fr